
Síndrome de Bart: reporte de caso.
Bart syndrome: case report.
Autor: 1.Medina Escalante Ricardo Arturo 1, 2., 2. Rodriguez Medina Ricardo 3.
1.Médico Residente de Tercer año Pediatría. Universidad Autónoma de Yucatán.
2. Hospital General de Cancún “Jesús Kumate Rodríguez”.
3. Intensivista Pediatra adscrito a Unidad de Terapia Intensiva Neonatal y Pediátrica, Hospital General Chetumal.
Correspondencia: Ricardo Rodríguez Medina
Esta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla.
RESUMEN
Introducción. El síndrome de Bart (SB) es una condición genética con defectos cutáneos al nacer, como falta de piel y ampollas. Puede heredarse y se relaciona con cambios en el colágeno tipo VII. Afecta principalmente extremidades, a veces con malformaciones renales. El diagnóstico se basa en evaluación clínica, pruebas genéticas y biopsias. El diagnóstico temprano es vital para prevenir complicaciones. Caso presentado en Hospital General de Chetumal.
Caso clínico. Recién nacido masculino con epidermolisis bullosa ingresó al Hospital General de Chetumal. Madre adolescente, infección urinaria en el embarazo. Trabajo de parto sin problemas. Presenta ausencia de piel en piernas y pies, distrofia ungueal y ampollas. Ingresó por dificultades respiratorias, con mejoría. Tratado con antibióticos. Análisis normales, diagnóstico de epidermólisis bullosa. Tratamiento tópico con mejoría importante. Alta a los 3 meses, reingreso a los 6 meses por infección de tejidos blancos, se recuperó sin complicaciones.
Conclusiones. El SB es poco conocido, su evolución varía y se basa entratamientotópico.Nohaytratamientoespecíficoenlaliteratura, es hereditario y requiere seguimiento familiar. Datos clínicos son vitales para el diagnóstico y un enfoque multidisciplinario para mejorar el pronóstico.
Palabras clave: “Síndrome de Bart” “Aplasia Cutis” “Epidermolisis” “Epidermolisis Bullosa”.
ABSTRACT
Introduction. Bart syndrome (BS) is a genetic condition characterized by cutaneous defects at birth, such as skin absence and blisters. It can be inherited and is linked to changes in collagen type VII. It mainly affects the limbs, sometimes with renal malformations. Diagnosis is based on clinical assessment, genetic testing, and biopsies. Early diagnosis is vital to prevent complications. Case presented at Chetumal General Hospital.
Clinical Case. A male newborn with epidermolysis bullosa was admitted to Hospital General de Chetumal. Teenage mother, urinary infection during pregnancy. Uncomplicated labor. The newborn had skin absence on legs and feet, nail dystrophy, and blisters. Admitted due to respiratory difficulties, improved. Treated with antibiotics. Normal analyses, diagnosed with epidermolysis bullosa. Treated topically with significant improvement. Discharged at 3 months, readmitted at 6 months due to tissue infection, recovered without complications.
Conclusions. Bart syndrome is little-known, with variable progression and treated topically. No specific treatment in the literature, hereditary, and requires family monitoring. Clinical data are crucial for diagnosis, and a multidisciplinary approach improves prognosis.
Keywords: “Bart Syndrome” “Aplasia Cutis” “Epidermolysis” “Epidermolysis Bullosa”.
INTRODUCCIÓN
El síndrome de Bart (SB) es una triada clínica de etiología genética, caracterizado por defectos cutáneos presentes al nacimiento, con patrón de herencia autosómico dominante y causada por alteraciones del colágeno tipo VII del cromosoma 3p(1). Expuesto por Bruce Bart en 1966, fue quien describió una triple asociación: ausencia localizada de piel (aplasia cutis), ampollas cutáneas (epidermolisis bullosa) y anomalías ungueales (ausencia congénita o distrofia ungueal)(2). Las lesiones de SB suelen presentarse en extremidades como úlceras rojas relucientes predominantemente unilaterales, afectando la superficie medial y / o dorsal de las extremidades.
Este padecimiento ocurre con frecuencia en solitario o asociado con anomalías del desarrollo como atresia pilórica, estenosis ureteral, anomalías renales, artrogriposis, oídos o nariz anormales y bandas amnióticas(3).
La aplasia cutis congénita (ACC) es un grupo de enfermedades heterogéneas, suele ser la característica inicial de SB pudiendo ser uni o bilateral, identificada siempre al nacimiento. Suele observarse en partes del cuerpo sujetas a alta fricción y trauma, siendo esta asociada a cualquier subtipo de epidermolisis bullosa (EB)(4).
Para 1986 se propuso por Frieden la clasificación de ACC en 9 grupos, dependiendo cada una de la ubicación de las lesiones y presencia de anomalías asociadas, la cual se encuentra vigente hasta el momento(5). Según esta clasificación, el grupo 6 se definió como SB contemplando la tríada antes citada.
No solo la genética juega un papel importante para este síndrome, sino que existen condiciones relacionadas tales como teratógenos específicos, traumas uterinos, compromiso vascular, infección intrauterina o movimientos fetales.
Aunque el SB suele ser diagnosticado clínicamente, el protocolo diagnóstico debe completarse con biopsia cutánea, estudio genético, inmunofluorescencia para evaluación histológica y microscopía electrónica para clasificar el subtipo de EB(6).
SB debe diagnosticarse precozmente para instituir el tratamiento adecuado, buscando prevenir complicaciones como infección local, sepsis, hemorragia, pérdida excesiva de fluidos por las lesiones, hipotermia, disturbios de electrolitos y, más tarde, cicatrices hipertróficas y atróficas(3).
Se presenta caso de paciente referido al área neonatal del Hospital General de Chetumal, con la finalidad de dar a conocer la evolución de un síndrome poco conocido y el manejo otorgado en la institución.
CASO CLÍNICO
Recién nacido, masculino, ingresado al Hospital General de Chetumal, producto de primera gesta, madre sana de catorce años, antecedente de infección urinaria activa al momento del parto. Control prenatal irregular con 3 consultas desde el primer trimestre. Embarazo normoevolutivo, sin exposición a radiación o tóxicos. Se desconocen antecedentes paternos, fuera del núcleo familiar, sin consanguinidad con madre. Parto a término vía vaginal, se realizan pasos iniciales de reanimación con adecuado esfuerzo respiratorio, llanto enérgico y adecuado tono de extremidades, APGAR 8/9, Silverman 0/0, Capurro 38sdg, Peso 2440 gramos, somatometría dentro rangos normales. A la exploración física, el paciente presentaba ausencia de tejido cutáneo en región pretibial de ambas piernas; además de tarso, metatarso, dorso y planta, 1o, 2o, 3o, 4o y 5o dedos de pie derecho. Se observa también distrofia ungueal en pie derecho y una lesión ampollosa en 4o falange mano izquierda. El resto de la exploración sin alteraciones. (figura 1 y 2).
FIGURA 1.
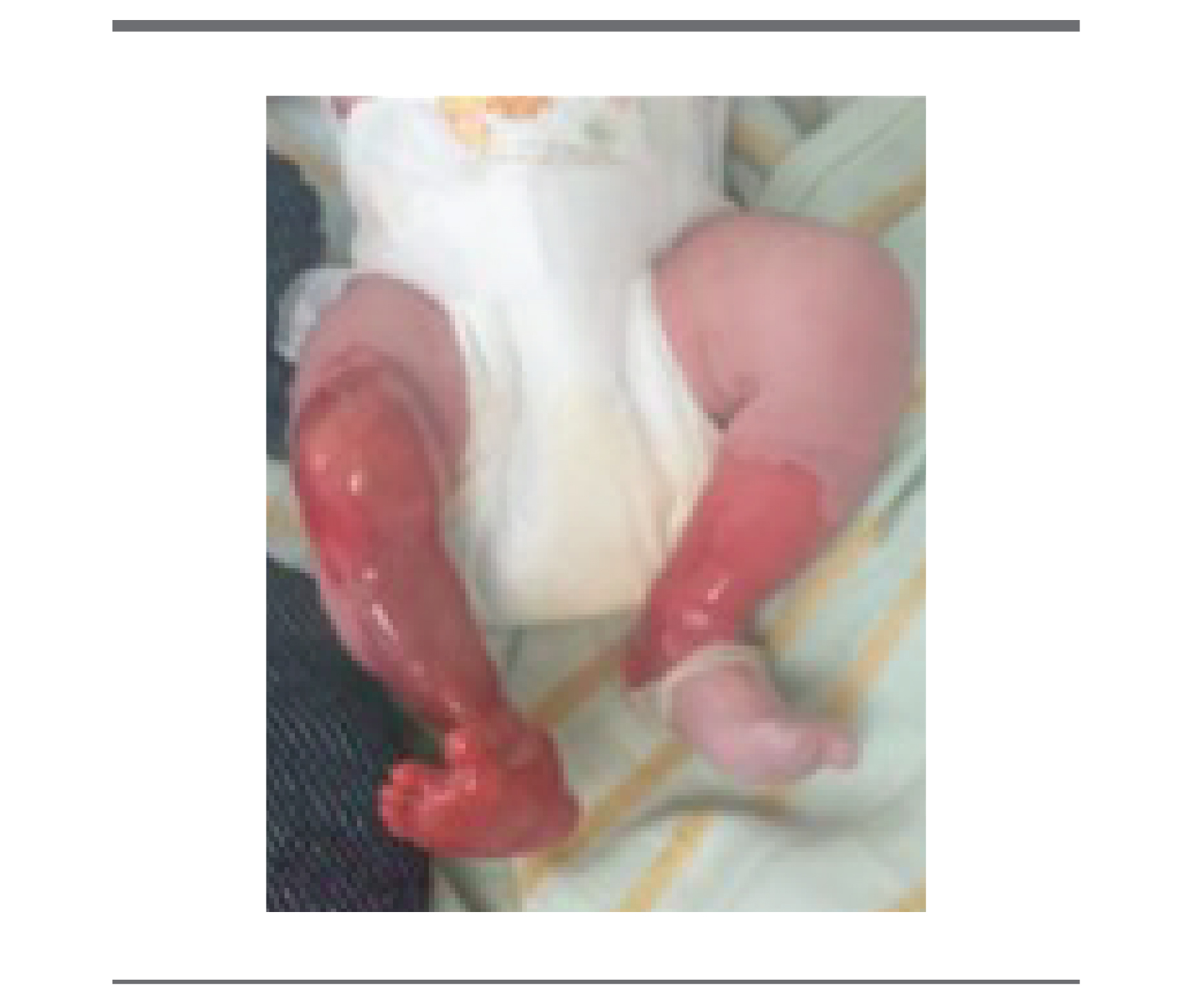
FIGURA 2.
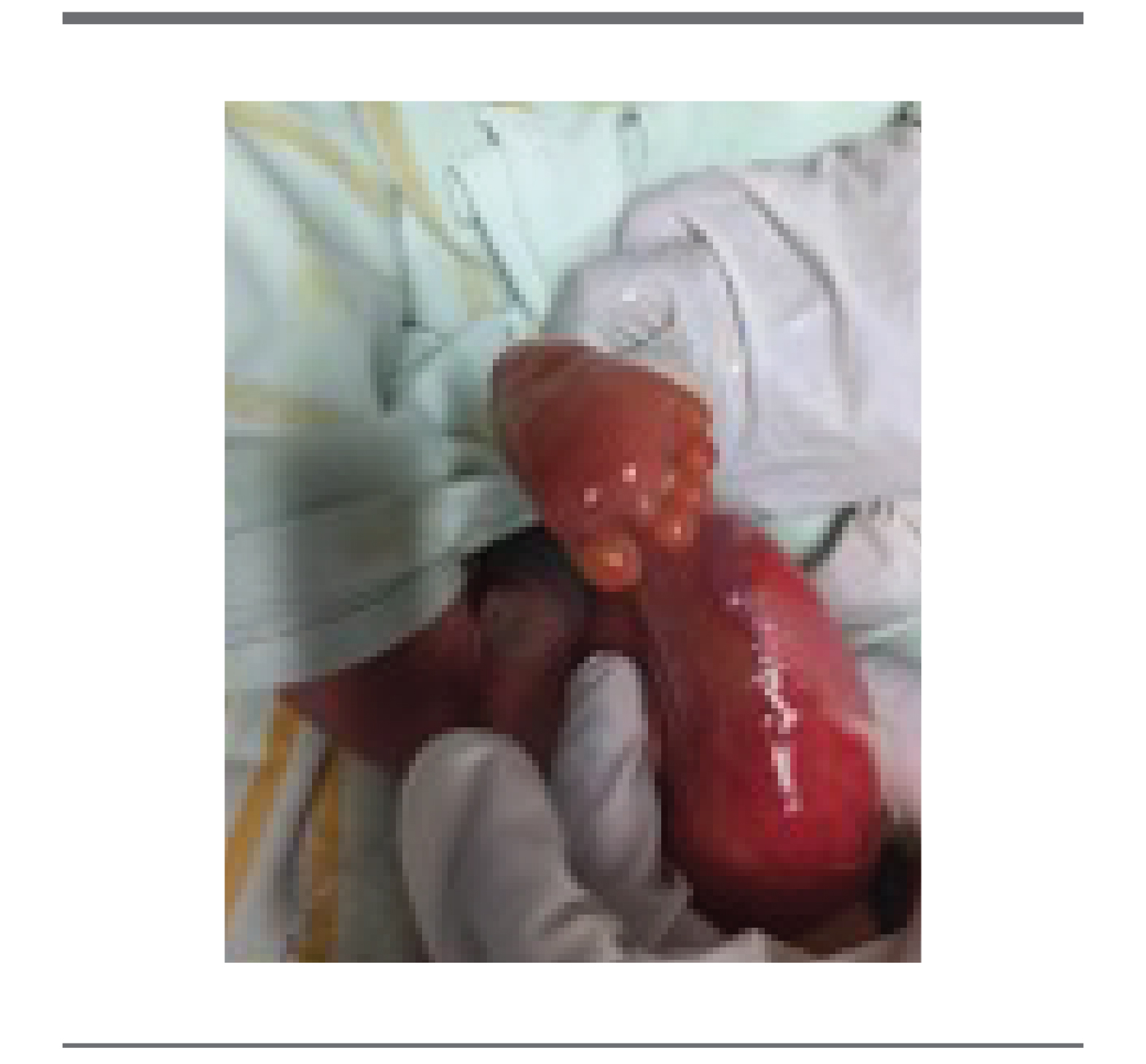
Ingresa a sala de cuidados intensivos neonatales por la presencia de las lesiones. Posterior al nacimiento con leve dificultad respiratoria, con apoyo ventilatorio con CPAP nasal durante 1 semana, posteriormente manteniendo con escafandra mejorando el patrón respiratorio y sin requerir nuevamente apoyo suplementario de oxígeno. Se iniciaron curaciones oclusivas con antibioticoterapia, se optó con tratamiento conservador tópico con mupirocina y sistémico ampicilina y amikacina;
Se tomó muestra del exudado de lesión cutánea para cultivo sin desarrollo bacteriano.
El hemograma, bioquímica sanguínea y pruebas metabólicas del paciente fueron normales. En la ecografía transfontanelar sin alteraciones. La ecografía renal y abdominal normal. El cariotipo del neonato: 46 XY, sin detectar anomalía cromosómica. Biopsia de piel con lesión subcutánea ampollosa compatible con epidermólisis bullosa. La tinción por inmunofluorescencia y microscopía electrónica necesarias para completar diagnóstico no están disponibles en nuestro medio hospitalario.
Se realizó valoración por cirugía plástica quien recomendó nuevo tratamiento tópico. Desde el momento en que las curaciones oclusivas semanales se realizaron con ketanserina gel 2%, parches mepilex, jelonet y gasa parafinada, se observó una notable mejoría de las lesiones. Durante estancia intrahospitalaria sin presentar foco séptico a ningún nivel, con adecuada evolución de lesiones sin sobreinfección de las mismas.
A las tres semanas de vida, y cuando en las zonas donde inicialmente existía una ausencia de piel, aparecía piel atrófica e hipopigmentada, se notó aparición de ampollas en área de piel previamente afectada o perilesional ante estímulos mecánicos que curaban con rapidez. La evolución fue insidiosa pero favorable a la postre, llevando adecuado tratamiento tópico, así como evitar trauma local y el contacto directo con superficies rugosas. (figura 3, 4 y 5).
FIGURA 3.
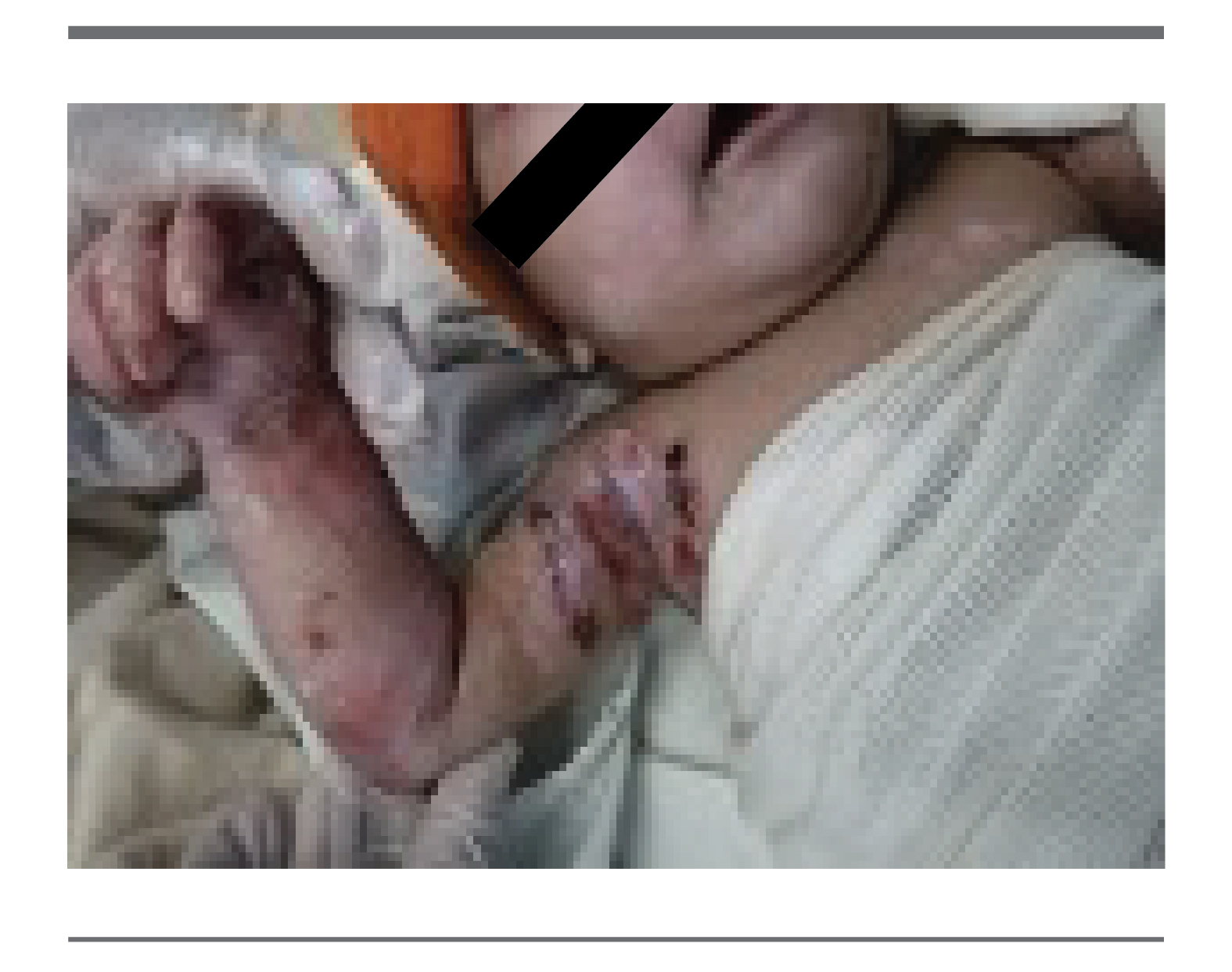
FIGURA 4.
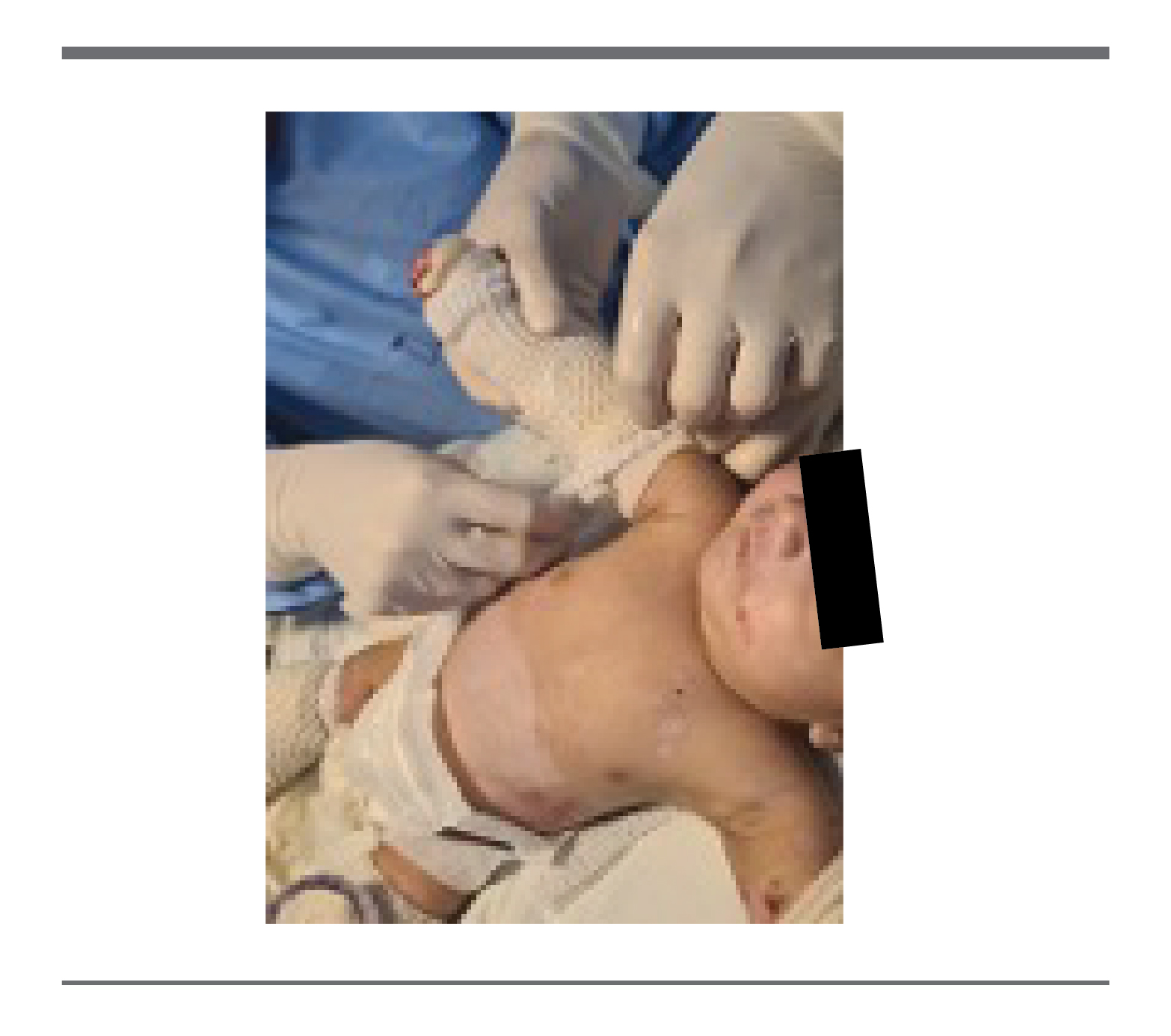
FIGURA 5.
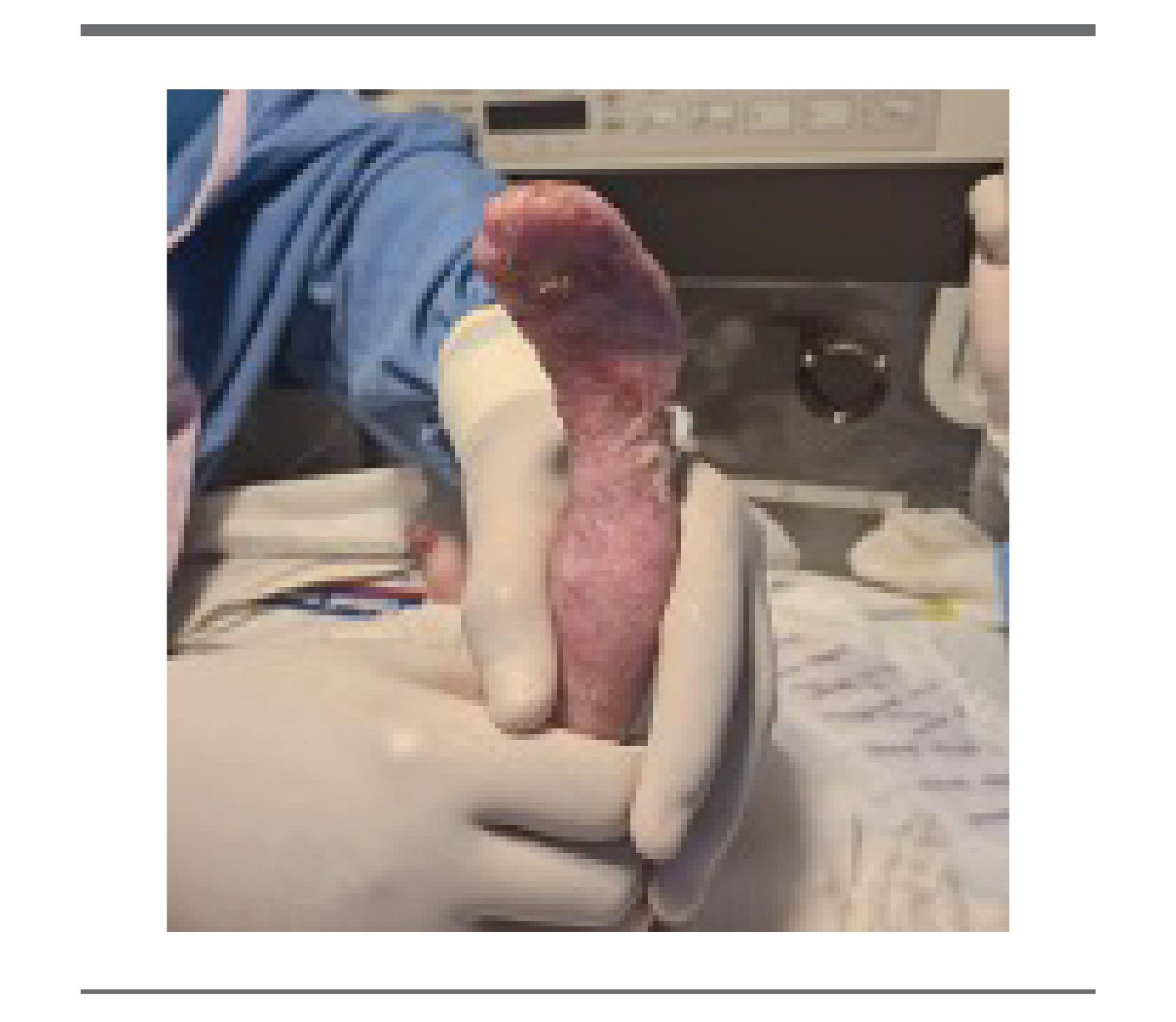
Se mantuvo hospitalizado con tratamiento tópico, dado de alta a los 3 meses de edad. Reingresado tres semanas después por cuadro de deshidratación e infección de tejidos blandos con germen aislado Pseudomonas spp, evolucionando satisfactoriamente con tratamiento sistémico y tópico, dado de alta nuevamente a los 6 meses de edad.
DISCUSIÓN
Paciente quien al nacimiento cursa con la asociación de ausencia congénita de piel en extremidades inferiores, epidermolisis bullosa por biopsia y anomalías ungueales que nos llevó al diagnóstico de Síndrome de Bart o aplasia cutis congénita tipo VI.
Durante estancia hospitalaria se realizaron estudios de extensión con el fin de poder descartar principales anomalías anatómicas asociados al SB.
Cursó con buena respuesta clínica de las lesiones aplicando exclusivamente tratamiento tópico, evitando los traumas locales y el contacto directo de las zonas afectadas con vendajes.
A los seis meses de vida la mayoría de las zonas que al nacimiento se presentaban desprovistas de piel, aparecían ahora, cubiertas por piel atrófica e hipopigmentada, quedando secuela en pie 97 derecho con sindactilia, en seguimiento por consulta externa para tratamiento de secuelas a corto y lego plazo.
CONCLUSIONES
El SB es una enfermedad poco conocida, cuya evolución es variable y depende exclusivamente del tratamiento tópico administrado. Actualmente la bibliografía no reporta tratamiento dirigido para esta enfermedad, que la vuelve de difícil control y suelen cursar con múltiples infecciones de tejidos blandos. No se debe olvidar la naturaleza de SB no es esporádica, sino hereditaria por lo que se debe tener un seguimiento estrecho a familiares, hasta el momento sin reporte de algún antecedente dentro del genograma.
Conocer datos clínicos de SB para el diagnóstico son fundamentales, pudiendo instaurar un tratamiento multidisciplinario para mejorar el pronóstico a corto y largo plazo.
REFERENCIAS
1. Reddy CU, Reddy KS, Reddy JJ. Bart syndrome: a rare entity. Arch Oral Res. 2011;7(1):69-73.
2. Bart BJ, Gorlin RJ, Anderson VE, Lynch FW. Congenital localized abscense of skin and associated abnormalities resembling epidermolysis bullosa: a new syndrome. Arch Dermatol. 1966; 93:296-304
3. Shahidi-Dadras M, Niknezhad N, Asadi-kani Z, Zaresharifi S, Hamedani B, Abdollahimajd F. Bart syndrome associated with skeletal deformities: An uncommon case report. Dermatologic Therapy. 2019;32:131.
4. Bajaj DR, Qureshi A. Bart's syndrome: a case report. J Pak Assoc Dermatol. 2008;18(2):113-5.
5. Frieden IJ. Aplasia cutis congenita: a clinical review and proposal for classification. J Am Acad Dermatol. 1986;14(4):646-660.
6. Albernaz RT, Silva AP. Síndrome de Bart: Relato de caso. Resid Pediatr. 2016;6(2):94-
7. Matthew H. Kanzler, MD, Bruce Smoller MD: Is Bart ́s Syndrome a Specific Syndrome. Arch Dermatol 1993; 129:907-908.




